792
Views & Citations10
Likes & Shares
The aging heart undergoes well characterized structural changes associated with functional decline, though the underlying molecular mechanisms remain obscure. Recent studies suggest that age-related increases in oxidative stress are associated with elevations in cardiac apoptosis and compensatory hypertrophy of the remaining cardiac myocytes. Although initially beneficial, prolonged cardiac hypertrophy can lead to decompensation, arrhythmias, contractile dysfunction and subsequent heart failure. Here we present the age-related molecular changes in the expression and phosphorylation of 4E-BP1, p-4E-BP1 (Thr 37/46), AKT, p-AKT (Thr 308), eEF2, p-eEF2 (Thr 56), P70s6K, p-P70s6K (Thr 421/Ser 424), S6rP, p-S6rP (Ser 235/236), STAT3, p-STAT3 (Ser 727), Tuberin/TSC2, p-Tuberin/TSC2 (Thr 1462), Calcineurin, eLF4E, and Raptor, in 6, 27, 30, 33 and/or 36 month F344XBN rat hearts underlying cardiac hypertrophy. This article represents data associated with prior publications from our lab. Taken together, these data suggest that very aged F344XBN rat hearts recruit several major signaling mechanisms to undergo hypertrophy.
Keywords: Age, Cardiac hypertrophy, F344XBN rat, Heart
INTRODUCTION
Cardiovascular disease is the leading cause of death for persons 65 years and older and is responsible for ~25% of deaths [1]. The aging heart is characterized structurally and functionally by increased hypertrophy, fibrosis, arrhythmias and diastolic dysfunction [2]. Previous studies have shown that age-associated cardiac alterations involve the extracellular signal-regulated kinase 1/2 (ERK1/2), protein kinase B (Akt) and the mammalian target of rapamycin (mTOR) signaling pathways [3-6]. These studies, however, did not use the National Institutes on Aging recommended aging rodent model, the Fischer 344/Norway F1 (F344/BNF1) [7]. The F344xBN rat undergoes aging similar to humans in which it exhibits increased left hypertrophy, fibrosis, and diastolic dysfunction [2,8]. mTOR signaling in the F344xBN rat has been investigated previously; however, the regulation and downstream signaling targets of mTOR and ERK 1/2 have not to our knowledge been elucidated in the aging F344xBN rat heart [8].
In previous studies which investigated cardiovascular and vascular disorders, ischemia/reperfusion injury, atherosclerosis and cardiovascular complications from diabetes, therapeutic strategies involving inhibiting or inducing Akt/mTOR signaling reduced or protected tissues [9,10]. A better understanding of the age-associated regulation in these signaling pathways could lead to therapeutic strategies targeting mTOR/Akt/ERK1/2 signaling for age-related pathologies such as hypertrophy.
EXPERIMENTAL DESIGN, MATERIALS AND METHODS
Animals
Animal care and procedures were conducted in accordance with the Animal Use Review Board of Marshall University using the criteria outlined by the American Association of Laboratory Animal Care (AALAC) as proclaimed in the Animal Welfare Act (PL89-544, PL91-979and PL94-279). Young (6 months, n=6), adult (27 months, n=6), aged (30-months, n=6) and very aged (33 and 36 months, n=6) male F344xBN rats were obtained from the National Institute on Aging. F344xBN rats were chosen for this study since this hybrid rat lives longer and has a lower rate of pathological conditions than inbred rats. The National Institutes on Aging probability of survival curves generated by indicate that the 30 months and 36 months rats used corresponded roughly to humans in their sixth and eighth decades of life, respectively. Rats were housed two per cage in an AALAC approved vivarium. Housing conditions consisted of a 12 h: 12 h dark-light cycle with temperature maintained at 22 ± 2°C. Animals were provided food and water ad libitum. Rats were allowed to acclimate to the housing facilities for at least 2 weeks before experimentation began. During this time, the animals were carefully observed and weighed weekly. None of the animals exhibited signs of failure to thrive, such as precipitous weight loss, disinterest in the environment or unexpected gait alterations.
Tissue collection
Rats were anesthetized with a ketamine-xylazine (4:1) cocktail (50 mg/kg, intraperitoneal) and supplemented as necessary for reflexive response before tissue collections. After midline laparotomy, the heart was removed and placed in Krebs-Ringer bicarbonate buffer (KRB) containing; 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24.2 mM NaHCO3 and 10 mM D-glucose (pH 7.4) equilibrated with 5% CO2/95% O2 and maintained at 37°C. Isolated hearts were quickly massaged to remove any blood from the ventricles, cleaned of connective tissue, weighed and immediately snap frozen in liquid nitrogen.
Tissue homogenization and determination of protein concentration
Cardiac muscles were homogenized in a Pierce Tissue Protein Extraction Reagent (T-PER) (10 mL/g tissue; Rockford, IL, USA) that contained protease inhibitors (P8340, Sigma-Aldrich, Inc., St. Louis, MO, USA) and phosphatase inhibitors (P5726, Sigma-Aldrich, Inc., St. Louis, MO, USA). After incubation on ice for 30 min, the homogenate was collected by centrifuging at 12,000 g for 5 min at 4°C. The protein concentration of homogenates was determined via the Bradford method (Fisher Scientific, Rockford, IL, USA). Homogenate samples were boiled in a 1X Laemmli sample buffer (Sigma-Aldrich, Inc., St. Louis, MO, USA) for 5 min.
SDS-PAGE and immunoblotting
50 µg of total protein from each sample (n=6) was separated on a 10% PAGEr Gold Precast gel (Lonza, Rockland, ME, USA) and then transferred to on a nitrocellulose membrane. Visual verification of transfer and equal protein loading amongst lanes was accomplished by Ponceau S staining of the membranes. For immunodetection, membranes were blocked for 1 h at room temperature in blocking buffer (5% non-fat dry milk in TBS-T (20 mM Tris-base, 150 mM NaCl, 0.05% Tween-20), pH 7.6), serially washed in TBS-T at room temperature, then probed with antibodies for the detection of 4E-BP1 (#9452), phosphor-4E-BP1 (Thr 37/46) (#9459), Akt (#9272), phospho-Akt (Thr308) (#9275), eEF2 (#2332), phospho-eEF2 (Thr56) (#2331), P70s6K (#9205), phosphor-P70s6K (Thr 421/ Ser 424) (#9204), S6rP (#2217), phospho-S6rP (Ser235/236) (#4858), STAT3 (#9132), phosphor-STAT3 (Ser 727) (#9145), Tuberin/TSC2 (#3612), phospho-Tuberin/TSC2 (Thr1462) (#3617), calcineurin (#2614), elF4E (#2067) and raptor (#2280) (from Cell Signaling Technology, Inc., Beverly, MA, USA). Membranes were incubated overnight at 4°C in primary antibody buffer (5% BSA in TBS-T, pH 7.6, primary antibody diluted 1:1000), followed by washing in TBS-T (3x 5 min each), and incubation with HRP-conjugated secondary antibody (anti-rabbit (#7074) or anti-mouse (#7076), Cell Signaling Technology, Inc., Danvers, MA, USA) in blocking buffer for 1 h. After removal of the secondary antibody, membranes were washed (3x 5 min each) in TBS-T and protein bands visualized on reaction with ECL reagent (Amersham ECL Western Blotting reagent RPN 2106, GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). The FluorChem E System (Cell Biosciences, Santa Clara, CA, USA) was used to detect immunoreactivity. Target protein levels were quantified by AlphaViewFC image analysis software (Alpha Innotech, San Leandro, CA, USA) utilizing the built in local background setting. Due to the digital nature of the image, pixel density information may not be apparent in reproductions of the original data files.
DATA ANALYSIS
Data were analyzed using Sigma Stat 12.0 statistical software and the results are presented as mean ± SEM. A one-way analysis of variance was performed for overall comparisons with the Student-Newman-Keuls post hoc test used to determine differences between groups. The level of significance accepted a priori was ≤ 0.05.
RESULTS
DISCUSSION
Cardiac hypertrophy involves multiple protein signaling pathways that are involved in cell growth, cell metabolism, protein synthesis and the cell cycle [11]. The regulation of cardiac hypertrophy is an area of active research however it is still unclear how this process is controlled at the molecular level [12,13]. Previous studies in our laboratory have shown in that age-associated cardiac hypertrophy, ROS-dependent smooth muscle hypertrophy and impaired overload-induced hypertrophy in insulin-resistant skeletal muscle was associated with alterations in ERK1/2, mTOR and Akt signaling [8]. To extend upon these observations, we examined the expression and activation (ratio of phosphorylated to total protein expression) of Akt, mTOR binding partner (Raptor), mTOR inhibitor (TSC2) and mTOR downstream targets (4E-BP1, p70s6k, rpS6, eEF2k, STAT3, Raptor and calcineurin) in the aging F344xBN heart.
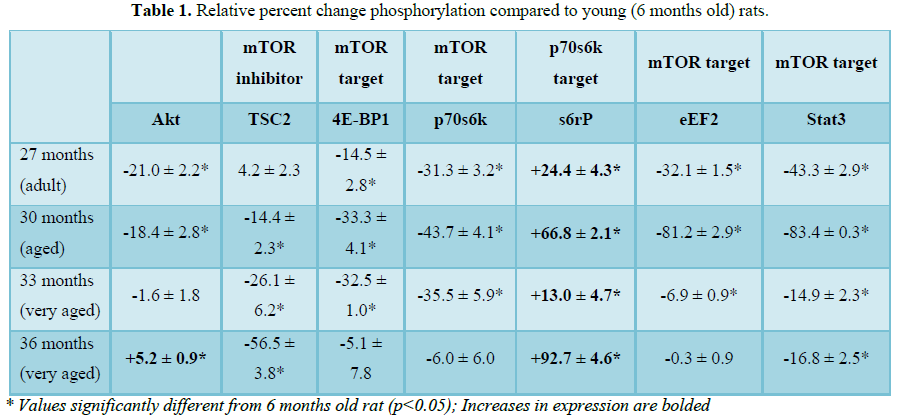
Previous findings from our laboratory reported an increase in Akt Ser473 phosphorylation at 36 months [8]. Activation of Akt requires phosphorylation of Ser473 and Thr308 [14]. In this study, Akt activation by phosphorylation of Thr308 was increased with age at 30, 33 and 36 months (Table 3 and Figure 1C). This result in addition to the increased activation of Akt Ser473 in our previous study confirms that activation of Akt was increased with age and may be responsible for the increased activation of its downstream target mTOR [14].
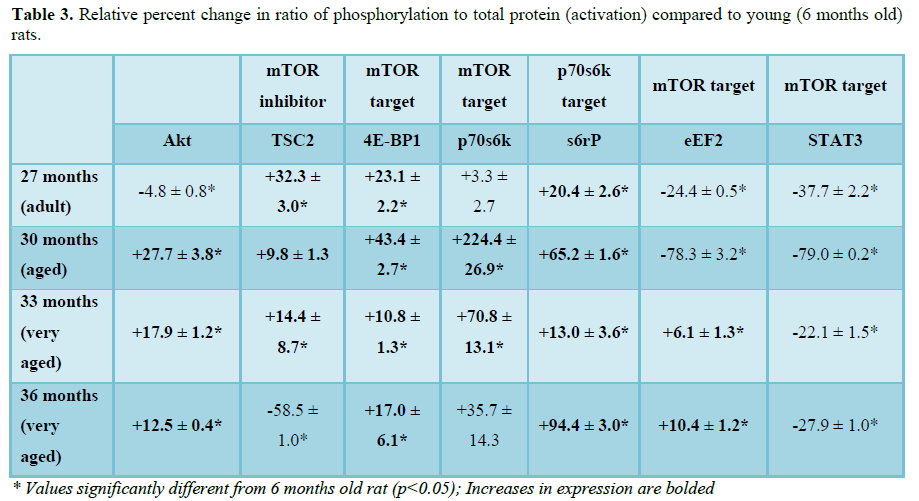
To determine how aging affects mTOR signaling, the expression and activation of the mTOR inhibitor (TSC2), mTOR binding partner (Raptor) and downstream targets were determined in the F344xBN heart. Previous work has shown a reduced inhibition of mTOR in hypertrophic states is associated with lower expression and activation of TSC2 [15-17]. In the aging F344xBN the activation of TSC2 was increased only slightly at 27 and 33 months. However, the expression of p-TSC2 was decreased from 27-33 months (Table 1 and Figure 3A) and the total TSC2 expression (Table 2 and Figure 2B) was decreased from 30-36 months which together, suggests a reduced activation of TSC2 (Table 3 and Figure 2C). Supporting these data, we found that the average heart weight of the 36 months old animals was nearly twice that of a 6 months old rat (Table 4) and significantly greater in mass than even the 30 months old heart [8].
mTOR signaling (phosphorylation) of the downstream targets (4E-BP1, p70s6 kinase) requires the binding of Raptor to mTOR [18-21]. Although, total expression of Raptor was decreased with aging (Table 2 and Figure 3C), the activation of 4E-BP1 was increased with aging (27-36 months) and was not affected by reduced Raptor expression (Table 3 and Figure 4C). This may have been due to both the decreased expression of mTOR inhibitor TSC2 (Table 2 and Figure 3B) and the trend of decreasing TSC2 activation over time from 27-36 months old (i.e., from +32.3% to -58.5% activation compared to 6 months old rats, respectively) (Table 3 and Figure 2C).
mTOR plays a critical role in cell growth by inhibiting 4E-BP1, an inhibitor of translation initiation by binding to eIF4E [22]. In the aging F344xBN heart there was a decrease in 4E-BP1 phosphorylation (27-33 months) (Table 1 and Figure 4A) and expression (27-36 months) (Table 2 and Figure 4B), but an increase in activation (27-33 months) (Table 3 and Figure 4C). The hyperphosphorylation of 4E-BP1 disrupts its interaction with eIF4E, which results in activation of cap-dependent translation [23].
While mTOR activation of p70s6k most likely occurred, the expression and activation of this protein can be confounded by expression of the structurally similar protein, p90s6k which is not activated by mTOR (Tables 1-3 and Figure 5) [24]. Increased mTOR induced p70s6k activation can, however, be detected by the increased activation of its target s6rP. S6rP, which is responsible for cell growth and cell cycle progression [25-28], was significantly activated compared to 6 months values at all-time points (Table 3 and Figure 6C).
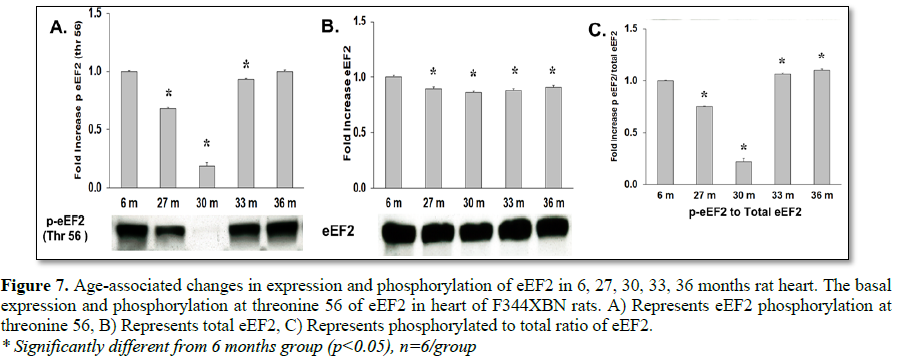
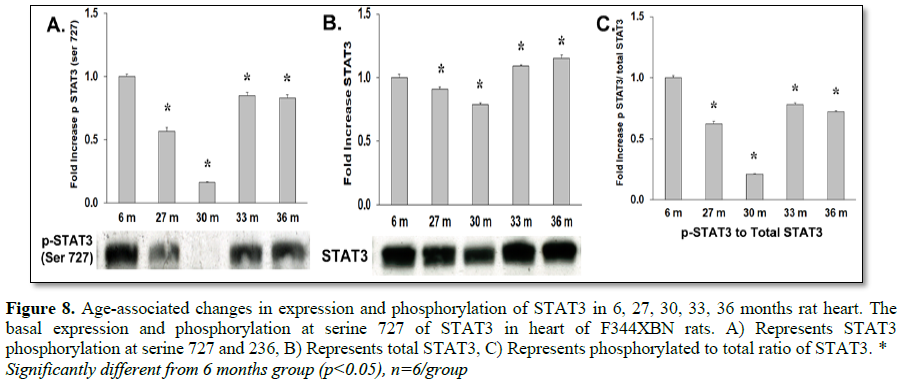
Eukaryotic elongation factor 2 kinase (eEF2k), another downstream target of mTOR, phosphorylates and inactivates eEF2 resulting in the inhibition of peptide-chain elongation [29]. This inhibition involves the phosphorylation of eEF2k by p90RSK and p70S6 kinase at Ser366 or by SAPK4/p38delta at Ser359 [30,31], which facilitates the dephosphorylation of eEF2 and thus promotes translation. In this study the downstream target of eEF2K, eEF2, was shown to have decreased activation at 30 months but not at 33 or 36 months (Table 3 and Figure 7C). Stat3, a transcription factor, is activated by mTOR pathways by the phosphorylation at Ser727 [32,33]. Expression and activation of Stat3 decreased in all age groups in the F344xBN (Tables 2 and 3 and Figures 8B and 8C).
Another important signaling molecule involved in cardiac hypertrophy is calcineurin, a phosphatase that is activated by calcium [34-36]. Other studies have shown that ERK1/2 and calcineurin often function together to induce cardiac hypertrophy [37]. Previous work in our laboratory has shown an increase in ERK1/2 signaling in the 36 months F344xBN heart. Here in, we demonstrate that the expression of calcineurin was significantly decreased with age (Figure 3A) in the F344xBN which is consistent with the possibility that this molecule may not play a major role in age-associated cardiac hypertrophy.
CONCLUSION
In summary, the findings from this study suggest that aging cardiac hypertrophy in the F344xBN rat is associated with the Akt activation of mTOR signaling pathways due to reduced expression of mTOR signaling inhibitor TSC2 and increased activation of downstream proteins involved in protein synthesis (p70s6k activated rpS6, and 4E-BP1). The activation of mTOR signaling could be due to the increased levels of ROS in the aging F344xBN heart [38]. Previous studies have shown increased mTOR activation with increased levels of ROS and the reduction of mTOR signaling with the use of antioxidants in cardiac pathologies [39-42]. Additional mechanistic studies involving the role of ROS in the activation of age-associated hypertrophy and activation of mTOR signaling are needed in order to understand the molecular signaling to determine potential therapeutic mechanisms for the elderly.
1. Heron M (2018) Deaths: Leading causes for 2016. In: N.V.S.R.D.o.V. Statisticls (Ed.) 77.
2. Walker EM Jr, Nillas MS, Mangiarua EI, Cansino S, Morrison RG, et al. (2006) Age-associated changes in hearts of male Fischer 344/Brown Norway F1 rats. Ann Clin Lab Sci 36: 427-438.
3. Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y (2008) The RAS/MAPK syndromes: Novel roles of the RAS pathway in human genetic disorders. Hum Mutat 29: 992-1006.
4. Yamazaki T, Tobe K, Hoh E, Maemura K, Kaida T, et al. (1993) Mechanical loading activates mitogen-activated protein kinase and S6 peptide kinase in cultured rat cardiac myocytes. J Biol Chem 268: 12069-12076.
5. Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, et al. (2011) Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol 106: 1173-1191.
6. Proud CG (2009) mTORC1 signalling and mRNA translation. Biochem Soc Trans 37: 227-231.
7. Lipman RD, Chrisp CE, Hazzard DG, Bronson RT (1996) Pathologic characterization of Brown Norway, Brown Norway × Fischer 344 and Fischer 344 × Brown Norway rats with relation to age. J Gerontol A Biol Sci Med Sci 51: B54-B59.
8. Manne N, Kakarla S, Arvapalli R, Rice K, Blough E (2014) Molecular mechanisms of age-related cardiac hypertrophy in the F344XBN rat model. J Clin Exp Cardiolog 5: 2155-9880.
9. Das A, Reis F, Maejima Y, Cai Z, Ren J (2017) mTOR signaling in cardiometabolic disease, cancer and aging. Oxid Med Cell Longev 2017: 6018675.
10. Filippone SM, Samidurai A, Roh SK, Cain CK, He J, et al. (2017) Reperfusion therapy with rapamycin attenuates myocardial infarction through activation of AKT and ERK. Oxid Med Cell Longev 2017: 4619720.
11. Wincewicz A, Sulkowski S (2017) Stat proteins as intracellular regulators of resistance to myocardial injury in the context of cardiac remodeling and targeting for therapy. Adv Clin Exp Med 26: 703-708.
12. da Rocha AL, Teixeira GR, Pinto AP, de Morais GP, Oliveira LDC, et al. (2018) Excessive training induces molecular signs of pathologic cardiac hypertrophy. J Cell Physiol 233: 8850-8861.
13. Nakamura M, Sadoshima J (2018) Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15: 387-407.
14. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, et al. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541-6551.
15. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151-162.
16. Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, et al. (2002) Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem 277: 30958-30967.
17. Noki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648-657.
18. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177-189.
19. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR et al. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163-175.
20. Beugnet A, Wang X, Proud CG (2003) Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1. J Biol Chem 278: 40717-40722.
21. Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278: 15461-15464.
22. Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294: 1942-1945.
23. Pause A, Belsham GJ, Gingras AC, Donzé O, Lin TA et al. (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature 371: 762-767.
24. El-Salem M, Raghunath PN, Marzec M, Wlodarski P, Tsai D, et al. (2007) Constitutive activation of mTOR signaling pathway in post-transplant lymphoproliferative disorders. Lab Invest 87: 29-39.
25. Pullen N, Thomas G (1997) The modular phosphorylation and activation of p70s6k. FEBS Lett 410: 78-82.
26. Dufner A, Thomas G (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 253: 100-109.
27. Peterson RT, Schreiber SL (1998) Translation control: connecting mitogens and the ribosome. Curr Biol 8: R248-R250.
28. Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, et al. (1997) Rapamycin suppresses 5'TOP mRNA translation through inhibition of p70s6k. EMBO J 16: 3693-3704.
29. Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, et al. (1997) Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc Natl Acad Sci U S A 94: 4884-4889.
30. Wang X1, Li W, Williams M, Terada N, Alessi DR, et al. (2001) Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370-4379.
31. Knebel A, Morrice N, Cohen P (2001) A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J 20: 4360-4369.
32. Wen Z, Zhong Z, Darnell JE Jr (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82: 241-250.
33. Yokogami K, Wakisaka S, Avruch J, Reeves SA (2000) Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 10: 47-50.
34. Flanagan WM, Corthésy B, Bram RJ, Crabtree GR (1991) Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 352: 803-807.
35. Clipstone NA, Crabtree GR (1992) Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357: 695-697.
36. Shibasaki F, Price ER, Milan D, McKeon F (1996) Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature 382: 370-373.
37. Sanna B, Bueno OF, Dai YS, Wilkins BJ, Molkentin JD (2005) Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol Cell Biol 25: 865-878.
38. Kakarla SK, Fannin JC, Keshavarzian S, Katta A, Paturi S (2010) Chronic acetaminophen attenuates age-associated increases in cardiac ROS and apoptosis in the Fischer Brown Norway rat. Basic Res Cardiol 105: 535-544.
39. Guimaraes DA, Dos Passos MA, Rizzi E, Pinheiro LC, Amaral JH, et al. (2018) Nitrite exerts antioxidant effects, inhibits the mTOR pathway and reverses hypertension-induced cardiac hypertrophy. Free Radic Biol Med 120: 25-32.
40. Calamaras TD, Lee C, Lan F, Ido Y, Siwik DA, et al. (2015) The lipid peroxidation product 4-hydroxy-trans-2-nonenal causes protein synthesis in cardiac myocytes via activated mTORC1-p70S6K-RPS6 signaling. Free Radic Biol Med 82: 137-146.
41. Li HL, Huang Y, Zhang CN, Liu G, Wei YS, et al. (2006) Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and -independent signal pathways. Free Radic Biol Med 40: 1756-1775.
42. Handayaningsih AE, Iguchi G, Fukuoka H, Nishizawa H, Takahashi M, et al. (2011) Reactive oxygen species play an essential role in IGF-I signaling and IGF-I-induced myocyte hypertrophy in C2C12 myocytes Endocrinology 152: 912-921.
-
 Table 1
Table 1 -
Table 2
-
Table 3
-
Table 4


QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Allergy Research (ISSN:2642-326X)
- Journal of Rheumatology Research (ISSN:2641-6999)
- Journal of Neurosurgery Imaging and Techniques (ISSN:2473-1943)
- Journal of Cancer Science and Treatment (ISSN:2641-7472)
- BioMed Research Journal (ISSN:2578-8892)
- Journal of Oral Health and Dentistry (ISSN: 2638-499X)
- International Journal of Radiography Imaging & Radiation Therapy (ISSN:2642-0392)


